nmer
A Constrained Combinatorial Framework for Efficient N-mer Analysis of Microbial Communities#
The analysis of microbial communities often requires examining combinations of taxa to capture higher-order relationships. However, the number of possible combinations increases rapidly with the number of taxa, making exhaustive enumeration computationally impractical. In this work, we present a hybrid N-mer generation framework that balances complete enumeration with practical limits on computational cost. The approach uses Gray code traversal to efficiently generate combinations, enabling full enumeration for samples with moderate numbers of taxa. For larger samples, where combinatorial growth is concentrated at intermediate values of k, the method introduces an adaptive cap that is applied only when the number of combinations becomes prohibitively large. Within these regions, combinations are prioritized using an abundance-based scoring scheme, favoring those supported by stronger quantitative signals. This design preserves full coverage of lower-order combinations while selectively reducing the number of high-order combinations that are often sparse and difficult to interpret. Applied to microbial abundance data, the framework maintains representative combinatorial structure while keeping runtime manageable. Overall, this work provides a scalable approach for exploring taxa combinations and a practical foundation for incorporating N-mer representations into downstream analyses.
Author Summary#
Microbial communities contain many different organisms that interact in complex ways. One way to study these interactions is to look at combinations of taxa that appear together across samples. However, as the number of taxa increases, the number of possible combinations grows extremely fast, making this type of analysis difficult to run in practice.
In this work, we developed an approach that allows us to generate these combinations efficiently while keeping the analysis manageable. For samples with a moderate number of taxa, the method produces all possible combinations. For larger samples, where the number of combinations becomes very large, it automatically limits only the most computationally expensive regions, focusing on combinations that are more likely to be meaningful.
This balance allows us to keep the analysis both thorough and practical. By taking into account how common each taxon is across samples, the method prioritizes combinations that are more likely to reflect real biological patterns. Overall, this approach provides a more scalable way to study how microbial communities are structured and how different taxa may interact.
Introduction#
Microbial communities are complex systems whose structure emerges from interactions among many taxa across environmental or host-associated contexts. Analyses focused on single taxa can identify dominant organisms or differential abundance patterns, but they often miss higher-order structure embedded in recurring combinations of taxa. Combinatorial representations can therefore provide a richer view of microbial community organization by capturing groups of taxa that appear together across samples.
The difficulty is computational. For a dataset containing n active taxa, the number of possible combinations rises according to the binomial coefficient and expands rapidly as k increases. Even at moderate values of n, exhaustive generation across many k values becomes impractical because both memory use and runtime grow quickly. In applied microbiome settings, this creates a tension between the biological appeal of higher-order combinations and the computational cost of enumerating them.
Several existing strategies address related problems but do not directly solve controlled taxa-level combination generation. Sequence-based k-mer methods are powerful for genomic and metagenomic classification, yet they operate on sequence fragments rather than combinations of taxa. Correlation and network-based methods infer associations among microbial features, but they do not explicitly enumerate combination space. This leaves an open methodological gap for approaches that can represent taxa combinations directly while remaining computationally tractable.
In practice, this challenge is not uniform across datasets. For samples with a limited number of active taxa, complete enumeration of combinations is feasible using efficient traversal strategies. However, as the number of taxa increases, the combinatorial space becomes concentrated around intermediate values of k, where the number of possible combinations grows most rapidly. In these regions, exhaustive generation can dominate computational cost while contributing combinations that are often sparse and difficult to interpret in biological terms.
The aim of this study was to develop a hybrid N-mer framework for taxa-level combinatorial analysis that adapts to varying levels of combinatorial complexity. Rather than relying on a fixed pre-generation restriction, the method performs full enumeration when computationally feasible and introduces adaptive constraints only in regions of the search space where combinatorial growth becomes prohibitive.
Specifically, this work introduces a k-dependent hybrid generation strategy that dynamically transitions from exhaustive enumeration to constrained sampling, guided by abundance-based scoring to prioritize combinations with stronger quantitative support. This design enables scalable exploration of taxa combinations while preserving biologically interpretable structure in the resulting N-mer representations. In this way, the framework provides a practical approach to managing combinatorial growth while retaining combinations that are more likely to reflect meaningful patterns in microbial community structure.
Materials and Methods#
Data Source#
Microbial abundance data used in this study were obtained from datasets generated at the Texas Children’s Microbiome Center. The data were organized as taxa-by-sample abundance matrices, where each entry represents the relative abundance of a given taxon within a sample.
These datasets were used to evaluate the N-mer generation pipeline under realistic conditions. They capture common characteristics of microbial communities, including variation in the number of taxa per sample and uneven abundance distributions.
Data representation#
For each sample, taxa with non-zero abundance values were considered active and included in combinatorial generation. The number of active taxa varied across samples, leading to differences in the size of the combinatorial search space.
Abundance values were retained and associated with each taxon throughout the analysis, allowing quantitative information to be incorporated into downstream scoring and prioritization of combinations.
N-mer generation using Gray code traversal#
Combinations of taxa (N-mers) were generated using a binary reflected Gray code traversal strategy. In this representation, each combination is encoded as a binary vector, where each position corresponds to a taxon and indicates its presence or absence in the combination.
Gray code ordering ensures that consecutive combinations differ by only a single element, allowing efficient traversal of the combinatorial space without repeated reconstruction of combinations. This reduces computational overhead and enables scalable enumeration for samples with a moderate number of taxa.
For samples with up to 28 active taxa, this approach enabled complete enumeration of all possible combinations across values of k. This threshold was determined empirically based on observed runtime and memory constraints in the current implementation.
Hybrid combinatorial generation strategy#
For samples with larger numbers of active taxa, full combinatorial enumeration becomes computationally impractical due to the rapid increase in the number of possible combinations. To address this, a hybrid generation strategy was applied in a k-dependent manner.
For each sample and each value of k, the number of possible combinations was evaluated using the binomial coefficient \(\binom{n}{k}\), where n is the number of active taxa. Exact enumeration was performed when the number of combinations remained below a predefined threshold of 40,116,600. When this threshold was exceeded, the corresponding k value was handled using the hybrid strategy.
Within these regions, the number of generated combinations was limited to a maximum of 3,000,000 per value of k. As a result, hybrid generation was not applied uniformly across all values of k, but instead activated selectively depending on the combinatorial complexity for each sample.
This approach allows full enumeration in tractable regions of the search space while controlling computational cost in regions where combinatorial growth becomes prohibitive.
Abundance-based scoring and selection#
Within capped regions, an abundance-based scoring scheme was used to prioritize combinations. For each combination, a score was calculated as the sum of the abundance values of its constituent taxa within a sample:
\[ S = \sum_{i=1}^{k} a_i \]where a_i represents the abundance of the i-th taxon in the combination. Combinations were ranked based on this score, and higher-scoring combinations were preferentially retained under the imposed cap. This approach favors combinations supported by stronger quantitative signals and reduces the contribution of combinations dominated by low-abundance taxa.
Computational environment#
All computations were performed on a CPU-based server environment at Baylor College of Medicine. The implementation was optimized for sequential and memory-efficient execution, leveraging Gray code traversal and conditional generation to manage combinatorial growth without reliance on GPU acceleration.
Evaluation of combinatorial growth#
The number of generated combinations was recorded for each value of k (i.e., for each N-mer size) to examine how the combinatorial space expands across different levels. For samples with up to 28 active taxa, full enumeration enabled direct observation of the complete combinatorial profile. For larger samples, the recorded counts reflect the imposed cap in high-density regions, capturing the effective behavior of the pipeline under practical constraints rather than the theoretical maximum number of combinations.
Evaluation strategy#
The performance of the proposed framework was evaluated in terms of computational scalability and the structure of the resulting combinatorial space. The analysis focused on how the number of generated combinations changes across values of k, the impact of adaptive capping on high-density regions, and the distribution of retained combinations under abundance-based prioritization. Dataset characteristics, including the number of active taxa per sample and taxa prevalence across samples, were also examined to provide context for the observed combinatorial behavior.
Results#
Dataset characteristics and taxa distribution#
The number of active taxa varied across samples, resulting in differences in the size of the combinatorial search space. Some samples contained relatively few active taxa, while others exhibited higher diversity, leading to substantially larger potential combination spaces. Taxa prevalence across samples showed a strongly skewed distribution. A small subset of taxa appeared consistently across many samples, whereas most taxa were observed infrequently. This uneven distribution contributed to the presence of many low-abundance and low-prevalence taxa, increasing the complexity of higher-order combinations. These prevalence patterns are shown in Fig 1.
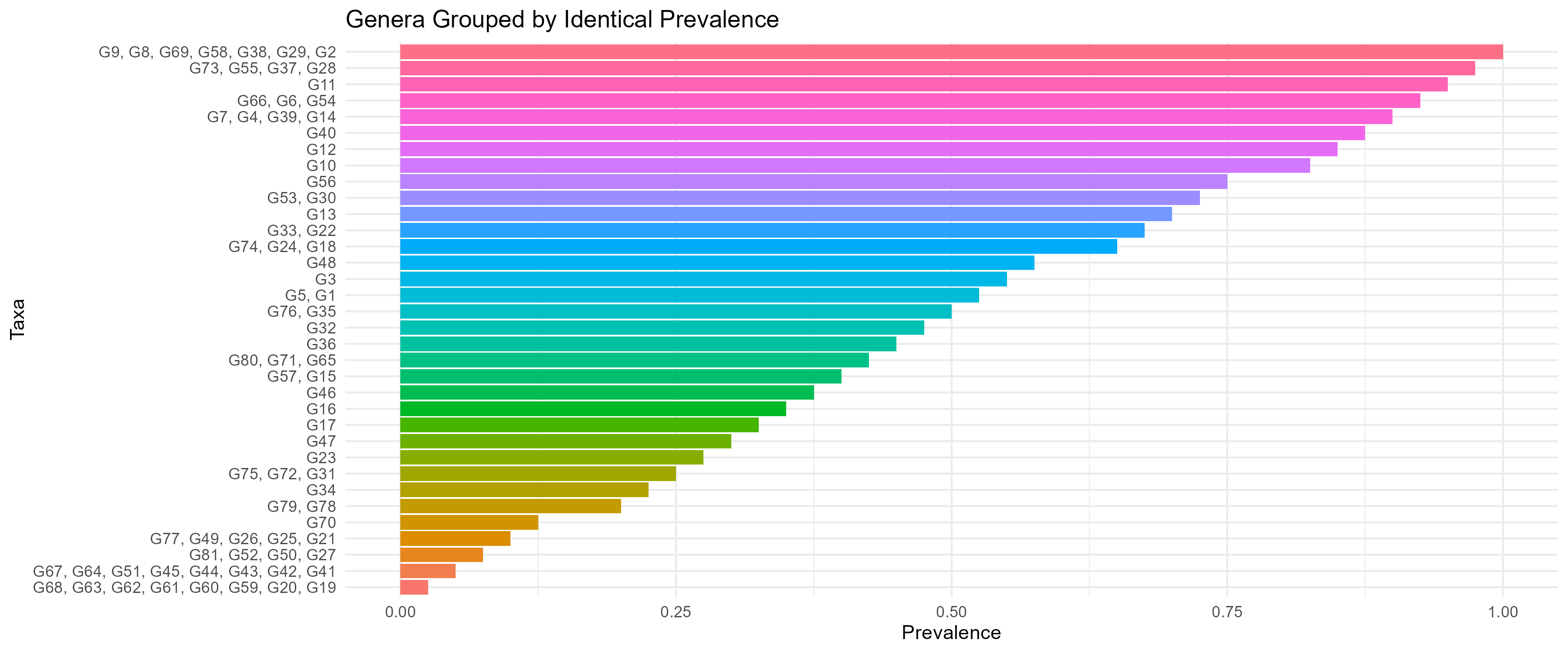
Figure 1: Distribution of taxa prevalence across samples Taxa are grouped based on the number of samples in which they are observed. The distribution shows a long tail of low-prevalence taxa.
Combinatorial profiles for fully enumerated samples#
For samples with smaller numbers of active taxa, the pipeline generated all possible combinations across values of k, resulting in complete combinatorial profiles.
To illustrate this behavior, we examined a representative sample with 28 active taxa. In this case, all combinations were generated, and the number of combinations increased with k, reaching a maximum of 40,116,600 at k = 14, before decreasing symmetrically as k approached the total number of taxa. This pattern reflects the full structure of the combinatorial space under exhaustive enumeration and demonstrates the pipeline’s ability to capture complete N-mer distributions when computationally feasible.
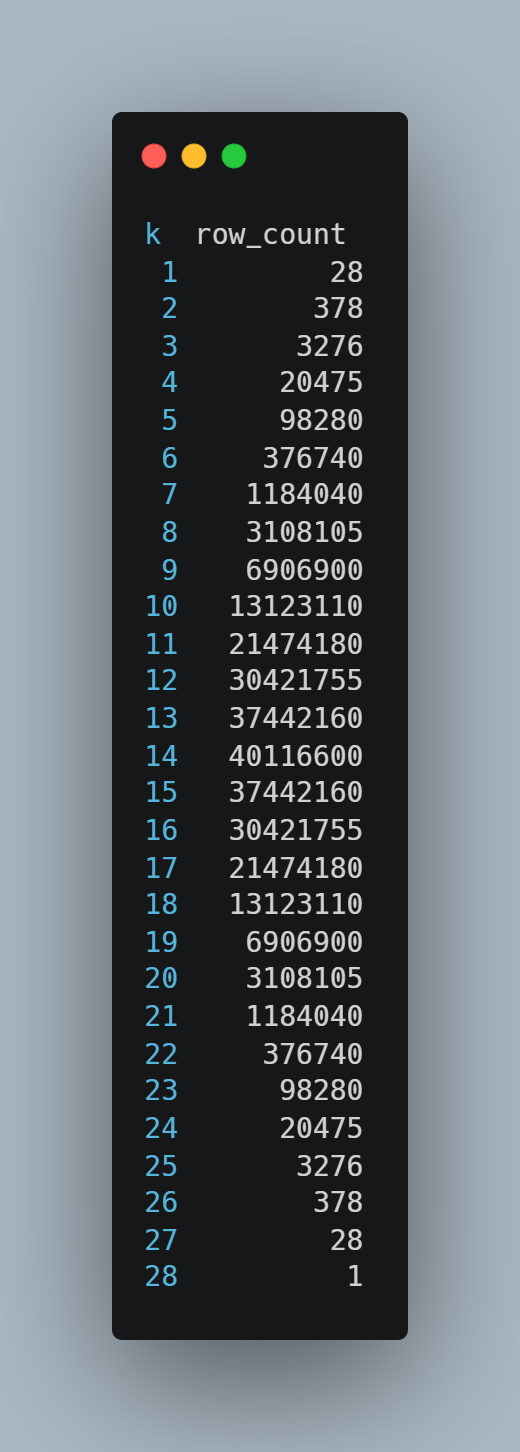
Figure 2: Number of generated combinations per N-mer level showing full enumeration across k.
Combinatorial profiles under hybrid generation#
For samples with larger numbers of active taxa, the transition from full enumeration to hybrid generation occurred at different values of k, depending on the combinatorial complexity of the sample. To illustrate this behavior, we examined a representative sample containing 39 active taxa. In this case, combinations were generated exhaustively for lower values of k, with the number of combinations increased from 39 at k = 1 to 15,380,937 at k = 7 values.
At k = 8, the number of possible combinations exceeded the predefined threshold, and the hybrid generation strategy was applied. From this point onward, the number of generated combinations reached the imposed cap of 3,000,000 per value of k, resulting in a plateau across a wide range of intermediate k values.
As k approached the total number of taxa, the number of combinations decreased again, reflecting the structure of the combinatorial space. This example demonstrates how the transition to hybrid generation is determined dynamically based on the number of active taxa and the resulting combinatorial growth.
Runtime behavior and computational scaling#
Execution time increased with both the number of active taxa and the overall combinatorial workload. For a representative sample with 28 active taxa, where full enumeration was performed, the pipeline completed in 59.6 seconds. This indicates that exact N-mer generation using Gray code traversal remains computationally feasible within this range.
For a sample with 39 active taxa, where hybrid generation was applied, runtime increased to 229.4 seconds. Despite the substantially larger combinatorial space, the increase in execution time remained controlled due to the selective application of the hybrid strategy.
When multiple samples were processed together, runtime scaled with the cumulative workload. For two samples (28 and 39 taxa), total execution time was 279.4 seconds, while for three samples (28, 27, and 39 taxa), runtime increased to 312.9 seconds. The observed increase was sublinear relative to the theoretical growth of the combinatorial space, reflecting the effect of capping in high-density regions. Across all runs, the majority of computation occurred during the primary generation stage, with negligible contributions from downstream processing steps.To further examine the effect of the hybrid selection threshold, we varied the hybrid_topk_per_k parameter across multiple values (3,000,000, 4,000,000, 5,000,000, and 10,000,000). Across the lower range of thresholds (3,000,000 to 5,000,000), total runtime remained relatively stable, ranging from approximately 312.9 to 321.3 seconds. This indicates that computational cost is primarily driven by combinatorial traversal rather than by the number of retained combinations within this range. However, when the threshold was increased to 10,000,000, runtime increased to 472.0 seconds, indicating that substantially larger caps begin to contribute to increased computational burden. Increasing the threshold beyond moderate values is therefore expected to expand the retained N-mer set while introducing additional combinations with lower abundance support, contributing limited interpretive value. These observations support the use of the selected cap as a practical balance between maintaining manageable runtime and avoiding unnecessary expansion of the output space.. Overall, these results demonstrate that the pipeline scales in a predictable and manageable manner, with the hybrid generation strategy effectively limiting excessive runtime growth in samples with large combinatorial spaces.
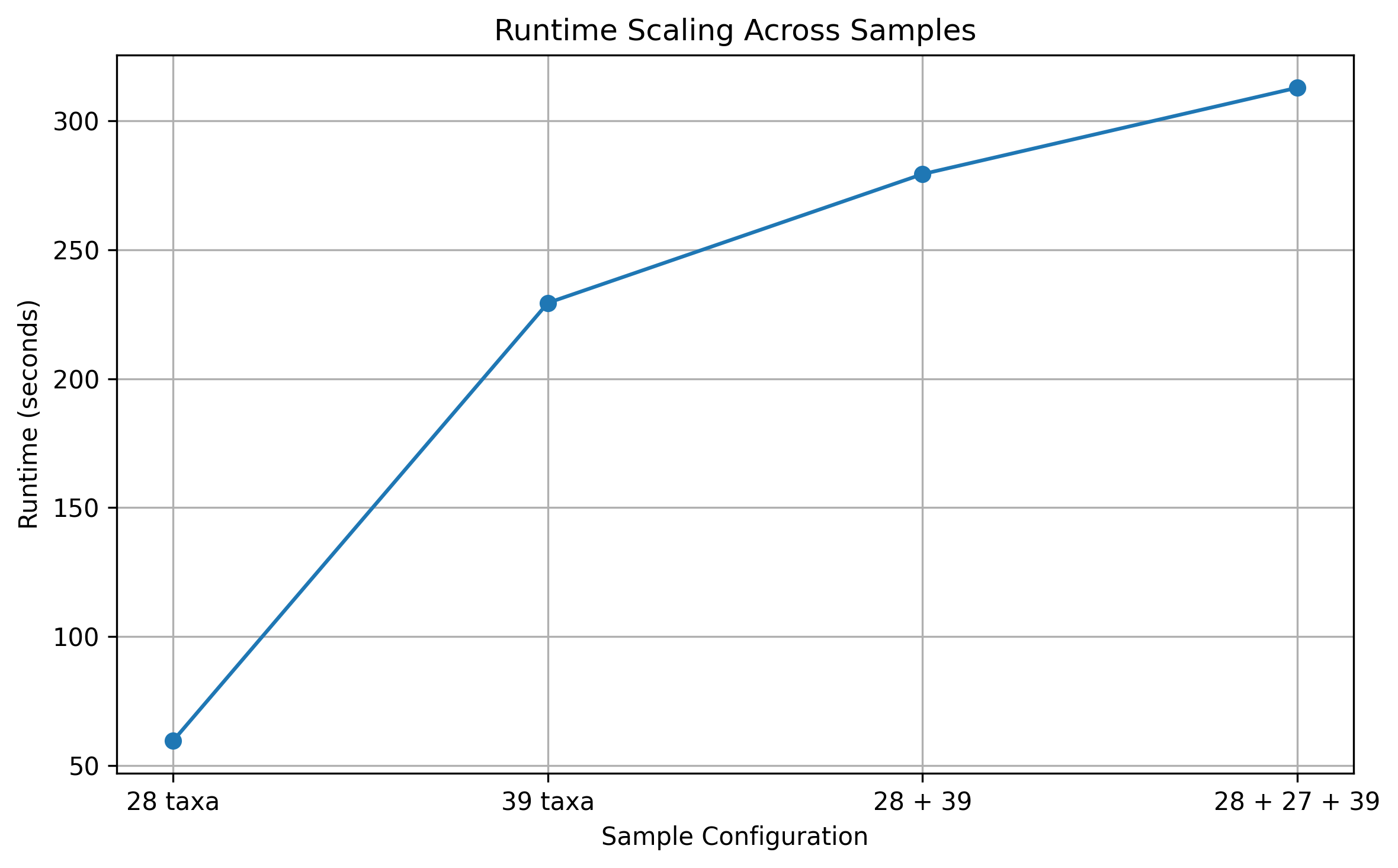
Figure 4: Execution time increased with sample size and combinatorial workload. The hybrid generation strategy limited excessive runtime growth in samples with larger combinatorial spaces.
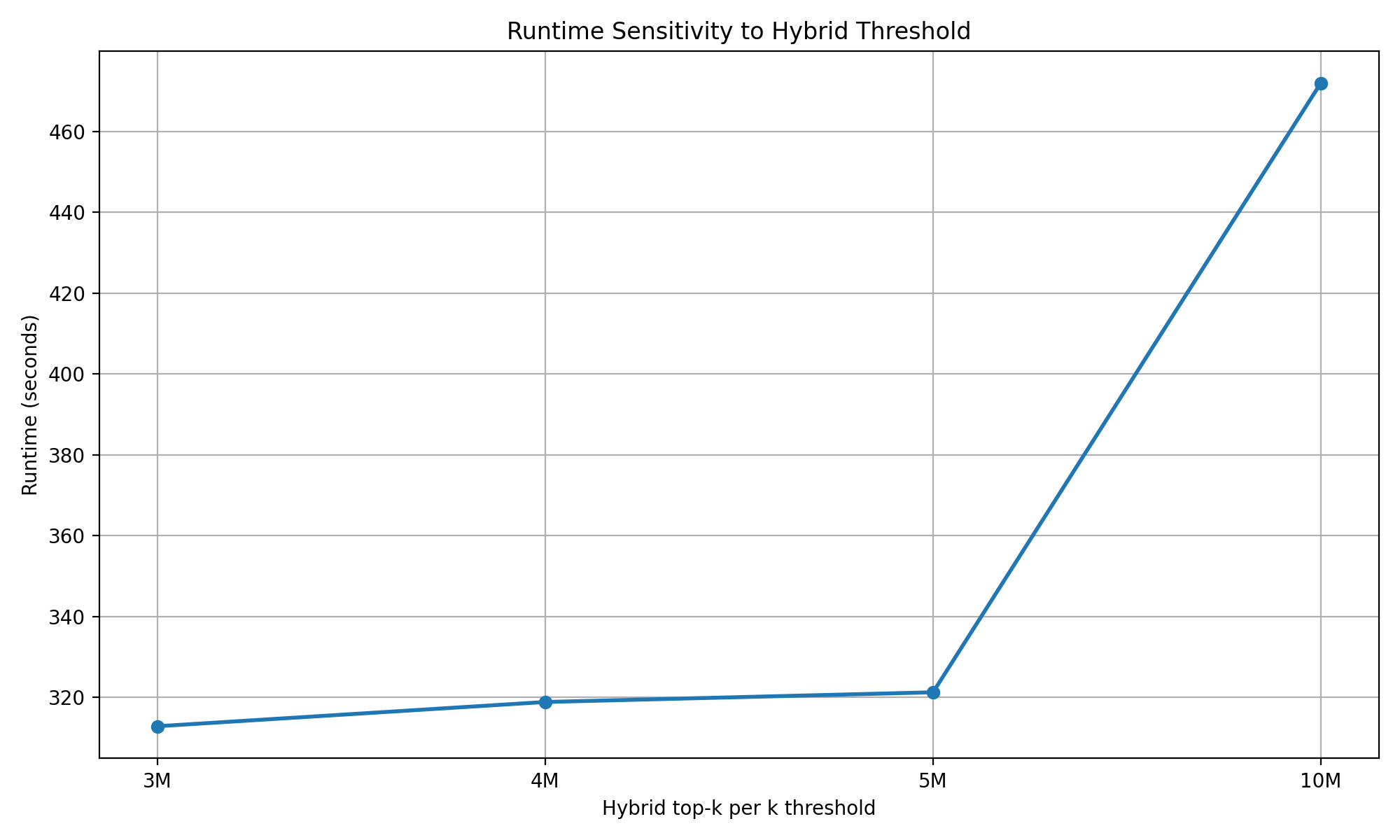
Figure 5: Execution time remained relatively stable as the hybrid selection threshold increased from 3,000,000 to 5,000,000 combinations per k, but increased when the threshold was raised to 10,000,000. This indicates that overall computational cost is primarily driven by combinatorial traversal within moderate threshold ranges, while larger caps begin to increase computational burden. The threshold primarily controls the size of the resulting N-mer set.
Discussion#
The results of this study highlight the central challenge of combinatorial explosion in taxa-based analyses. As the number of active taxa increases, the number of possible combinations grows rapidly, with the highest density occurring at intermediate values of k. While Gray code traversal enables efficient enumeration for smaller samples, full combinatorial generation becomes computationally impractical as the number of taxa increases.
The hybrid framework introduced here addresses this limitation by combining exhaustive enumeration with adaptive constraints. Rather than applying a fixed cutoff, the method adjusts dynamically based on the combinatorial complexity of each sample. As observed in the results, fully enumerated samples (e.g., 28 taxa) exhibit the expected symmetric combinatorial profile, whereas larger samples transition into a capped regime at sample-specific values of k. This transition leads to a characteristic plateau in the number of generated combinations, reflecting controlled exploration of high-density regions of the search space. A key strength of this approach is its selective control of combinatorial growth. Lower-order combinations are preserved through full enumeration, while higher-order regions are constrained without being entirely excluded. This allows the pipeline to maintain coverage of biologically interpretable combinations while avoiding excessive computational cost. The observed runtime behavior further supports this design, with execution time remaining manageable even for samples with substantially larger combinatorial spaces.
Within capped regions, abundance-based scoring was used to prioritize combinations with stronger quantitative support. This reduces the contribution of combinations dominated by low-abundance taxa, which are more likely to produce sparse and unstable patterns. Given the skewed structure of microbial datasets, this strategy helps focus the analysis on combinations that are more likely to reflect recurring community structure. Results from the threshold sensitivity analysis further support this design choice. While increasing the cap from 3,000,000 to 5,000,000 had limited impact on overall runtime, a larger increase to 10,000,000 led to a noticeable increase in computational cost. Increasing the threshold expands the number of retained combinations but is expected to introduce additional low-abundance combinations with reduced interpretive value. In the representative sample with 39 active taxa, the selected threshold provided sufficient coverage of high-scoring combinations within the capped region while maintaining manageable computational cost. These observations reinforce the role of the cap as a mechanism for controlling output complexity while preserving biologically relevant structure.
Despite these advantages, several limitations should be considered. First, adaptive capping introduces a trade-off between completeness and computational feasibility, and some higher-order combinations may be excluded. Second, the current framework relies primarily on abundance-based criteria, which do not directly account for consistency across samples. As previous work has shown, data preprocessing choices can significantly influence downstream results, and similar considerations apply to combinatorial analyses.
An important extension of this framework would be the incorporation of prevalence-based filtering prior to combinatorial generation. Low-prevalence taxa can inflate the number of active taxa without contributing consistent patterns, thereby expanding the search space with limited interpretive value. Reducing this noise would improve both computational efficiency and biological relevance. More broadly, integrating prevalence with abundance-based scoring could provide a more balanced approach to prioritizing informative combinations. While abundance captures within-sample signal strength, prevalence reflects reproducibility across samples. Combining these measures may improve the stability of features used in downstream analyses, including machine learning applications.
In summary, this study demonstrates that a hybrid, k-dependent generation strategy can effectively address the combinatorial explosion inherent in taxa-level analysis. By combining full enumeration in tractable regions with adaptive constraints in high-density regions, the framework maintains coverage of biologically interpretable combinations while controlling computational cost. This balance between completeness and efficiency allows higher-order taxa relationships to be explored without overwhelming computational resources. More broadly, the approach provides a practical foundation for incorporating N-mer representations into downstream statistical and machine learning analyses, where scalability and interpretability are both critical.
Acknowledgments#
The author would like to thank Dr. Nilesh Dixit and Dr. Sandra Cole from Arizona State University for their valuable discussions, guidance, and support throughout this work.
Code Availability#
The code used in this study is available from the authors upon reasonable request.